DeepMind publica una nueva base de datos pública de estructuras de proteínas predichas por IA que podría transformar la biología
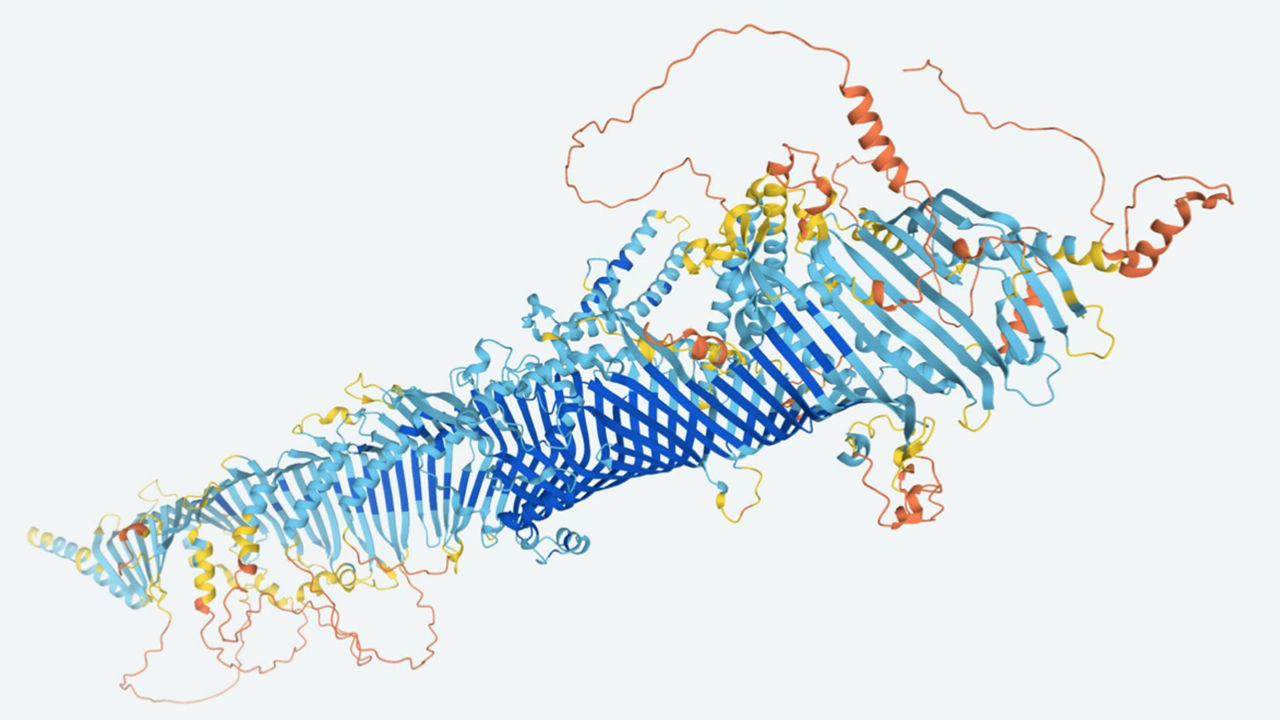 |
| Las computadoras ahora pueden predecir de manera rápida y confiable la forma 3D de la mayoría de las proteínas, como esta estructura de una mosca de la fruta. DEEP MIND |
Julio 23, 2021. DeepMind por fin consigue descifrar cómo están plegadas las proteínas, los ladrillos con los que se construye la vida. Cada proteina es una cadena de aminoácidos, como cuentas de perlas ensartadas que conforman el collar, y predecir su estructura es uno de los santos griales de la biología
DeepMind ha anunciado que su plan es publicar en los próximos meses 100 millones de estas estructuras, algo que hubiera costado miles de millones de años de investigación al ritmo convencional. "Es bastante abrumador", dice John Moult, un experto en plegamiento de proteínas de la Universidad de Maryland, Shady Grove, que dirige una competencia bienal llamada Evaluación crítica de la predicción de la estructura de las proteínas (CASP). Moult dice que los biólogos estructurales han soñado durante décadas que los modelos informáticos precisos algún día aumentarían las formas de proteínas extremadamente precisas derivadas de métodos experimentales como la cristalografía de rayos X. “Nunca pensé que el sueño se haría realidad”, dice Moult.
Según cuenta Science, el modelo de computadora, llamado AlphaFold, es el trabajo de investigadores de DeepMind, una empresa de inteligencia artificial del Reino Unido propiedad de Alphabet, la empresa matriz de Google. En el otoño de 2020, AlphaFold arrasó en la competencia CASP, con una puntuación de precisión media de 92,4 sobre 100, muy por delante del siguiente competidor más cercano. Pero debido a que los investigadores de DeepMind no revelaron los detalles de cómo mapearon teóricamente las formas de las proteínas, específicamente el código informático subyacente de AlphaFold, otros equipos se sintieron frustrados, incapaces de aprovechar el progreso. Eso empezó a cambiar la semana pasada.
AlphaFold determinó que muchas de las otras proteínas humanas estaban "desordenadas", lo que significa que su forma no adopta una estructura única. Tales proteínas desordenadas pueden finalmente adoptar una estructura cuando se unen a una proteína asociada, dice Baker. También pueden adoptar de forma natural múltiples conformaciones, dice David Agard, biólogo estructural de la Universidad de California en San Francisco.
Cada proteina es una cadena de aminoácidos, como cuentas de perlas ensartadas que conforman el collar, y predecir su estructura es uno de los santos griales de la biología: pues ciertas enfermedades, como el alzhéimer, el párkinson, la diabetes o la fibrosis quística, se originan en la acumulación de proteínas mal plegadas.
Debido a que la estructura 3D de una proteína dicta en gran medida su función, la biblioteca DeepMind es apta para ayudar a los biólogos a determinar cómo funcionan miles de proteínas desconocidas. "En EMBL creemos que esto será transformador para comprender cómo funciona la vida", dice la directora general del laboratorio, Edith Heard.
Los colaboradores de DeepMind dicen que AlphaFold2 ya ha estimulado el desarrollo de enzimas novedosas que descomponen los plásticos en el medio ambiente más rápidamente que las encontradas anteriormente y condujeron a nuevas posibilidades de medicamentos para tratar enfermedades desatendidas. "Este será uno de los conjuntos de datos más importantes desde el mapeo del genoma humano", dice Ewan Birney, director del Instituto Europeo de Bioinformática de EMBL. Así habla de este programa, AlphaFold, la directora general del Laboratorio Europeo de Biología Molecular, Edith Heard:
"Ha sido entrenado utilizando datos de recursos públicos creados por la comunidad científica, así que tiene sentido que sus predicciones sean públicas. Es una auténtica revolución para las ciencias de la vida, como lo fue la genómica hace décadas”.
No es probable que los impactos se detengan ahí. Las predicciones ayudarán a los experimentadores que resuelven estructuras, dice Baek. Los datos de los experimentos de cristalografía de rayos X y microscopía crioelectrónica pueden ser difíciles de interpretar, dicen Baek y otros, y tener un modelo puede ayudar. “A corto plazo, impulsará los esfuerzos de determinación de la estructura”, predice. "Y con el tiempo también reemplazará lentamente los esfuerzos de determinación estructural [experimental]". Si eso sucede, los biólogos estructurales no se quedarán sin trabajo.



Comentarios
Publicar un comentario